
Descriptions of Clinical and Surgical Procedures
Surrénalectomie rétropéritonéoscopique postérieure bilatérale avec épargne corticale du côté droit
Manuscript Format: Full Text
Main Text
Table of Contents
Abstrait
L’adrénalectomie avec conservation corticale permet la résection de la ou des tumeurs surrénales tout en préservant le tissu surrénalien non affecté pour prévenir l’insuffisance surrénalienne. Ceci est particulièrement important chez les patients atteints de tumeurs bilatérales des surrénales, généralement des phéochromocytomes.
La rétropérito-néostomie postérieure (PRA) permet une approche peu invasive de la résection de la glande surrénale par rapport à la surrénalectomie transabdominale laparoscopique plus traditionnelle et aux approches ouvertes. La technique PRA est de plus en plus utilisée par les chirurgiens endocriniens à haut volume dans le monde entier. Cette approche est idéale pour traiter les patients atteints d’une maladie bilatérale et a été utilisée dans ce cas d’un patient présentant des phéochromocytomes bilatéraux dans le cadre d’un syndrome de néoplasie endocrinienne multiple 2A.
Aperçu du cas
Arrière-plan
La rétropérito-néoscapomie postérieure (PRA) a été popularisée pour la première fois en Allemagne par Walz et ses collègues. Les glandes surrénales sont accessibles par une approche rétropéritonéale à l’aide d’instruments laparoscopique et d’insufflation de CO2 . 1 Ce faisant, le chirurgien évite l’entrée dans la cavité péritonéale et la mobilisation des viscères environnants, notamment l’intestin, le foie, la rate et le pancréas. Par rapport aux approches ouvertes et laparoscopique de surrénalectomie transabdominale (LTA), cette technique favorise le rétablissement du patient avec une durée de séjour réduite, moins de douleur et un risque réduit d’iléus. 2-4
L’un des avantages de la PRA est qu’elle permet un accès bilatéral aux deux glandes surrénales via une approche peu invasive sans nécessiter de repositionnement du patient pendant les opérations. 2 Les patients présentant des tumeurs bilatérales des surrénales, généralement des phéochromocytomes dus au syndrome de Von Hippel Lindau (VHL) ou au syndrome de néoplasie endocrinienne multiple de type 2 (NEM2), sont des candidats idéaux pour cette approche. Dans les deux processus pathologiques, des tumeurs bilatérales apparaissent fréquemment. En tant que tels, les patients peuvent avoir besoin d’une surrénalectomie bilatérale pour obtenir une guérison biochimique.
Afin de prévenir l’insuffisance surrénale aiguë postopératoire (crise addisonienne), une surrénalectomie corticale épargnant peut être réalisée. Au cours de cette procédure, le tissu tumoral coupable est retiré tout en préservant le tissu surrénalien normal. 5 Traditionnellement, cette technique a été décrite parallèlement aux approches ouvertes et LTA ; cependant, pour les maladies bilatérales, l’approche PRA est de plus en plus utilisée avec succès.
Antécédents ciblés du patient
Il s’agit d’une femme de 31 ans qui présentait des phéochromocytomes bilatéraux sans équivoque sur le plan biochimique. Elle présentait des symptômes d’hypertension et de palpitations, ce qui a incité son médecin traitant à approfondir ses recherches. Ses analyses de laboratoire ont révélé une élévation des métanéphrines plasmatiques libres à 642 pg/ml (plage de référence < 57 pg/mL) et de la normétanéphrine à 2284 pg/ml (référence < 148 pg/ml), ainsi qu’une élévation des métanéphrines urinaires, compatible avec un phéochromocytome. L’imagerie en coupe transversale comprenait une TDM de l’abdomen avec contraste IV. La TDM a révélé des nodules surrénaliens bilatéraux avec des unités de Hounsfield précontrastées et des caractéristiques d’imagerie globales du phéochromocytome bilatéral (Figure 1). Un examen attentif de son imagerie a révélé que le tissu du cortex surrénalien du côté droit était d’apparence normale (figure 2) et qu’il serait propice à une surrénalectomie épargnant le cortex du côté droit.
En raison de son jeune âge et de la présence de tumeurs bilatérales, elle a fait l’objet d’autres recherches pour d’autres tumeurs associées à la NEM2A, en particulier le cancer médullaire de la thyroïde et l’hyperparathyroïdie primaire. On a en effet constaté qu’elle avait un taux élevé de calcitonine sérique de 229 pg/ml (référence < 5 pg/ml), et les résultats de l’échographie et de l’aspiration à l’aiguille fine étaient compatibles avec un carcinome médullaire de la thyroïde du lobe thyroïdien droit. Son sérum intact, ses taux d’hormone parathyroïdienne et de calcium n’étaient pas remarquables.
Etudes d’imagerie
La TDM et l’IRM sont les principales techniques radiologiques utilisées pour l’imagerie des glandes surrénales normales et anormales. Sur la TDM, les phéochromocytomes sont souvent bien définis à partir des tissus environnants et présentent généralement des unités de Hounsfield pré-contrastées de 30 à 40. Les lésions plus petites ont tendance à sembler simples et solides, tandis que les lésions plus grandes peuvent avoir des caractéristiques plus kystiques ainsi que d’autres caractéristiques complexes. À l’IRM, les phéochromocytomes ont une apparence classique d’ampoule sur les images pondérées en T2. L’imagerie fonctionnelle peut également être obtenue, en particulier lorsque la maladie métastatique est préoccupante. La méthode la plus courante est l’utilisation de 131 I- et 123 I-métaiodobenzylguanidine (MIBG), qui est un analogue de la noradrénaline et se localise préférentiellement dans les tissus sympathomédullaires. 6
Nous préférons que le patient subisse une tomodensitométrie ou une IRM dans les 3 à 6 mois suivant l’opération pour la planification opératoire. Dans ce cas, le patient nous a été référé après avoir subi un scanner montrant des masses surrénaliennes bilatérales avec des caractéristiques d’imagerie suspectes de phéochromocytome. Lors d’un examen détaillé de son imagerie, il était évident que le membre médial inférieur de la glande surrénale droite avait un tissu du cortex surrénalien non affecté qui pourrait potentiellement être préservé lors de la résection.
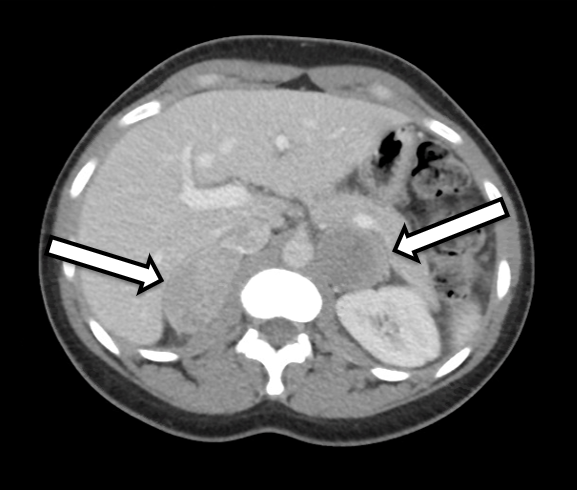
Graphique 1. Vue transversale du nodule à l’intérieur du lobe thyroïdien droit ;
Les flèches blanches indiquent des microcalcifications.
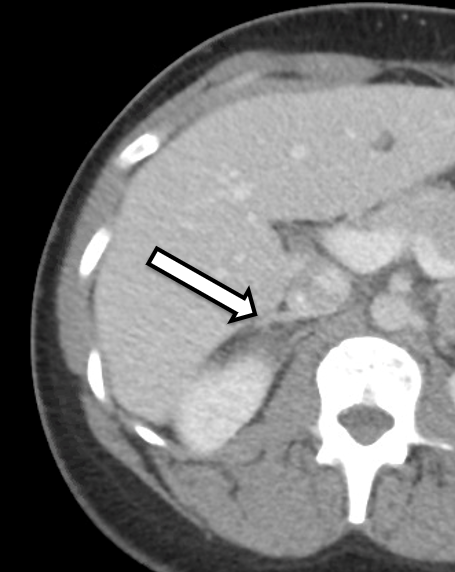
Graphique 2. Vue améliorée de la TDM de l’abdomen montrant
cortex surrénalien normal (flèche) de la surrénale droite
glande qui a été préservée.
Histoire naturelle
Les phéochromocytomes proviennent des cellules de la crête neurale de la médullosurrénale et sécrètent des quantités excessives de catécholamines. La prévalence globale des phéochromocytomes a été estimée entre 1:6500 et 1:2500, avec un âge moyen d’apparition entre 40 et 50 ans et une prévalence légèrement plus élevée chez les femmes. La plupart des tumeurs sont bénignes. 6 Il est intéressant de noter qu’il n’existe pas de caractéristique pathologique unique, y compris la taille, le taux mitotique ou l’invasion vasculaire ou capsulaire, qui puisse prédire avec précision le potentiel malin, bien que divers algorithmes prédictifs aient été créés. Environ 10 à 15 % des tumeurs peuvent présenter et/ou développer une maladie métastatique, indiquant une transformation maligne. 8 La survie spécifique à la maladie pour les phéochromocytomes malins a été estimée à environ 70 % à 5 ans, d’après l’analyse de la base de données SEER (Surveillance, Epidemiology, and End Results). 9
Options de traitement
Le traitement standard des phéochromocytomes est la résection chirurgicale. Avant l’opération, le patient doit subir un blocage des catécholamines, qui est généralement effectué à l’aide de la phénoxybenzamine, un bloqueur non sélectif des récepteurs alpha, ou d’un alpha-bloquant sélectif tel que la doxazosine. Des agents antihypertenseurs supplémentaires peuvent également être nécessaires, y compris des bêta-bloquants. Cependant, les bêta-bloquants ne doivent être commencés qu’après qu’un patient a été placé sous alpha-bloquants pour prévenir une crise hypertensive causée par un blocage non opposé des récepteurs bêta. De plus, les patients sont chroniquement déshydratés et nécessitent une réanimation liquidienne préopératoire. 10
Justification du traitement
Les patients atteints de phéochromocytomes souffrent fréquemment d’épisodes d’hypertension sévère et d’autres manifestations cliniques d’une production excessive de catécholamines, notamment des palpitations, des maux de tête, des attaques de panique et une diaphorèse. 11 Bien que les médicaments contre l’hypertension puissent apporter un soulagement partiel, le seul traitement durable à long terme est la résection de la ou des lésions coupables. De plus, ceux qui continuent sans intervention sont à risque de crises d’hypertension graves, pouvant entraîner la mort. 12 Par conséquent, les patients atteints de phéochromocytomes doivent rechercher un traitement chirurgical rapidement.
Chez les patients présentant des phéochromocytomes bilatéraux, l’adrénalectomie avec conservation corticale peut prévenir l’apparition d’une insuffisance surrénalienne tout en réséquant le tissu coupable. Des études antérieures ont montré que la surrénalectomie totale peut être très morbide. Par exemple, dans une série publiée par Lairmore et al. dans laquelle 43 patients ont subi une surrénalectomie complète pour des phéochromocytomes bilatéraux, 23 % ont souffert d’épisodes d’insuffisance surrénalienne et un patient est décédé d’une crise addisonienne. 13 De plus, les patients ayant subi une surrénalectomie totale font état d’une mauvaise qualité de vie et d’hospitalisations fréquentes liées à l’insuffisance surrénalienne. 14
Discussion
L’adrénalectomie avec conservation corticale est une option inestimable pour les patients atteints de syndromes génétiques associés à des phéochromocytomes. Ces syndromes comprennent la NEM2, le VHL et la neurofibromatose de type 1 (NF1), ainsi que d’autres. Il est important de noter que les patients diagnostiqués avec des phéochromocytomes bilatéraux doivent être orientés vers des tests génétiques. Le test de la calcitonine peut être un complément utile pour identifier les patients atteints de NEM2, car les tests génétiques peuvent prendre plusieurs mois pour être effectués et correctement interprétés. Environ 40 à 80 % des patients atteints de NEM2A ou de VHL développeront des phéochromocytomes bilatéraux, et ces tumeurs sont généralement bénignes. 15 En tant que telle, la surrénalectomie avec conservation corticale peut enlever les tissus affectés, tout en laissant suffisamment de tissu pour prévenir l’insuffisance surrénalienne avec un risque minimal de récidive.
Le premier cas d’adrénalectomie avec conservation corticale a été décrit par Irvin et al. en 1983. 16 Depuis, certains établissements ont signalé avoir réussi à pratiquer une surrénalectomie épargnant le cortex au moyen d’approches ouvertes ou peu invasives, avec des taux de récidive assez faibles et, surtout, une faible incidence d’insuffisance surrénalienne. 14 L’utilisation de l’ARP a augmenté à mesure que les chirurgiens endocriniens à haut volume ont mis en œuvre cette technique. L’ARP a été décrite pour la première fois en 1995, puis développée grâce à l’expérience de Walz et de ses collègues. 1, 4, 17 Des études rétrospectives comparant la PRA à la LTA montrent une diminution des temps opératoires, une diminution de la perte de sang et aucune différence dans les résultats à long terme.
Plus récemment, la PRA est également utilisée pour la surrénalectomie qui épargne le cortex. Dans une série récente, l’expérience sur une période de 25 ans de la réalisation d’adrénalectomies épargnant le cortex pour les phéochromocytomes bilatéraux à l’aide de l’approche RPA a été décrite. Soixante-six patients ont été opérés avec un total de 101 surrénalectomies effectuées. Leur temps opératoire moyen était de 128 minutes pour les chirurgies bilatérales, et ils n’ont signalé que deux complications majeures. Ils ont pu effectuer une opération d’épargne corticale dans 89 des cas et parmi ces patients, 91 % n’ont pas eu besoin de stéroïdes en postopératoire. Un seul patient présentait une maladie persistante, mais aucune récidive n’a été signalée. 18 Les patients doivent être avertis qu’ils pourraient souffrir d’insuffisance surrénalienne à la suite d’une intervention chirurgicale. Cela dépend de la taille du tissu surrénalien restant et de la préservation du flux sanguin. L’insuffisance surrénale temporaire est prise en charge en étroite consultation avec l’endocrinologie et les tests de laboratoire appropriés.
Anesthésie
L’intervention est réalisée sous anesthésie générale avec intubation endotrachéale. En raison des altérations hémodynamiques qui peuvent survenir pendant la chirurgie en raison de la tumeur, une ligne artérielle est placée pour la surveillance de la pression artérielle et l’anesthésiste administre des médicaments vasoactifs au besoin. Un cathéter de Foley est placé pour surveiller le débit urinaire. En fonction du degré d’élévation des catécholamines et d’autres facteurs du patient, un cathéter veineux central peut être placé pour un accès veineux supplémentaire et la surveillance de la pression veineuse centrale.
Technique chirurgicale
Après l’induction de l’anesthésie générale et l’intubation endotrachéale, le patient est placé en position couchée avec les hanches pliées à 90 degrés. Une table Cloward avec une selle chirurgicale Cloward est utilisée pour permettre à l’abdomen de pendre de manière dépendante. La position jackknife et la selle chirurgicale Cloward ouvrent l’espace rétropéritonéal pour maximiser l’exposition. Tous les points de pression, y compris le visage, les bras, les hanches et les jambes, sont largement rembourrés. La crête iliaque, les extrémités des 11e et 12e côtes et le bord des muscles paraépineux sont des points de repère importants marqués par le chirurgien. L’incision initiale est placée juste en dessous de l’extrémité de la 12e côte. Les ciseaux sont utilisés pour diviser nettement les tissus mous et pénétrer dans le rétropéritoine. Le doigt du chirurgien est ensuite utilisé pour dégager brutalement l’espace et guider le placement d’un port de 5 mm médialement et latéralement, tous deux inclinés à environ 30 degrés et dirigés vers la position de la glande surrénale. Un port de ballon de 10 mm est ensuite placé dans l’incision médiane. Le rétropéritoine est insufflé avec du CO2 à travers une tubulure à haut débit jusqu’à une pression d’insufflation de 25 mmHg.
Une lunette de 5 mm à 30 degrés est insérée dans le port de 10 mm, et un appareil LigaSure est utilisé pour créer l’espace rétropéritonéal. La dissection commence par l’identification médiale des muscles paraépineux, suivie de l’identification du rein inférieur. La caméra est ensuite généralement déplacée vers le port médial et le chirurgien utilise un appareil LigaSure et une pince intestinale à travers les ports latéraux et centraux, respectivement. La dissection se poursuit sur le pôle supérieur du rein et le long des muscles paraspinaux médialement, vers la direction de la glande surrénale. L’exposition est facilitée en partie par la pression vers le bas sur les reins. Au fur et à mesure que la glande surrénale est identifiée, l’étendue inférieure de la glande et sa relation avec la veine surrénale sont déterminées. À droite, cette dissection révèle la VCI, à partir de laquelle la glande surrénale est disséquée pour révéler la veine surrénale. À gauche, la veine surrénale provient de la veine rénale gauche.
À ce stade, la relation précise entre le tissu cortical surrénalien non altéré et la tumeur coupable est déterminée. Ceci est en partie déterminé par une évaluation détaillée de l’imagerie préopératoire en conjonction avec les résultats peropératoires. Parfois, l’échographie peropératoire peut être utile. Une fois qu’un plan approprié est identifié, le phéochromocytome est ensuite séparé du tissu surrénalien normal à l’aide d’un dispositif LigaSure. La glande surrénale est très vascularisée, de sorte qu’une attention méticuleuse est accordée à l’hémostase lors de la traversée de la glande. Dans ce cas, le membre médial inférieur de la glande surrénale droite native était approprié pour la préservation. Si possible, la veine doit être préservée. Cependant, ce n’est pas nécessaire, ni toujours faisable, étant donné qu’il y a un drainage veineux supplémentaire le long des petites artères surrénales. Au cours de l’opération actuelle, la veine surrénale a été trouvée pénétrant directement dans la tumeur et, en tant que telle, elle a été divisée avec des clips. À ce stade, les attaches médiales et latérales de la tumeur sont enlevées, laissant l’attache supérieure pour permettre à la tumeur de pendre et de fournir un site supérieur de contre-traction. On prend soin de ne pas pénétrer dans la cavité péritonéale lors des dissections latérales et céphalales. Une fois que la tumeur a été disséquée circonférentiellement, les attaches supérieures sont prélevées et la glande est retirée de l’espace rétropéritonéal avec un appareil Endocatch. Le port de 10 mm est fermé en couches, tandis que les sites de port de 5 mm ne sont fermés qu’au niveau de la peau.
Pathologie et suivi
La pathologie finale a révélé un phéochromocytome droit de 5,2 cm et un phéochromocytome gauche de 5,6 cm. Les tests effectués le premier jour postopératoire ont montré une légère diminution de la production de cortisol. En tant que tel, le patient a été temporairement placé sous une faible dose de stéroïdes oraux. La patiente a reçu son congé de la maison le deuxième jour postopératoire et s’est très rétablie lorsqu’elle a été vue à la clinique deux semaines après son opération, avec un plan pour se sevrer de la faible dose de prednisone.
Équipement
cadre Andrew, selle chirurgicale Cloward, dispositif LigaSure et sac de récupération Endocatch.
Divulgations
Les auteurs n’ont aucune divulgation.
Déclaration de consentement
Le patient visé dans cet article vidéo a donné son consentement éclairé pour être filmé et est conscient que des informations et des images seront publiées en ligne.
References
- Walz MK, Peitgen K, Hoermann R, Giebler RM, Mann K, Eigler FW. La rétropéritonéoscopie postérieure comme nouvelle approche mini-invasive pour la surrénalectomie : résultats de 30 surrénalectomies chez 27 patients. Monde J Surg. 1996; 20(7):769-774. doi :10.1007/s002689900117.
- Callender GG, Kennamer DL, Grubbs EG, Lee JE, Evans DB, Perrier ND. Rétropéritonéoscopie postérieure à la surrénalectomie. Adv Surg. 2009; 43(1):147-157. doi :10.1016/j.yasu.2009.02.017.
- Perrier ND, Kennamer DL, Bao R, et al. Surrénalectomie rétropéritonéoscopique postérieure : technique privilégiée pour l’ablation des tumeurs bénignes et des métastases isolées. Ann Surg. 2008; 248(4):666-674. doi :10.1097/SLA.0b013e31818a1d2a.
- Walz MK, Peitgen K, Walz MV, et al. Rétropérito-néoscopique postérieure : leçons apprises en cinq ans. Monde J Surg. 2001; 25(6):728-734. doi :10.1007/s00268-001-0023-6.
- Lee JE, Curley SA, Gagel RF, Evans DB, Hickey RC. Surrénalectomie avec conservation corticale chez les patients atteints de phéochromocytome bilatéral. Chirurgie. 1996; 120(6):1064-1071. doi :10.1016/S0039-6060(96)80056-0.
- Farrugia FA, Martikos G, Tzanetis P, et al. Phéochromocytome, diagnostic et traitement : revue de la littérature. Endocr Réguler. 2017; 51(3):168-181. doi :10.1515/ENR-2017-0018.
- Thompson LD. Phéochromocytome de la glande surrénale Scaled Score (PASS) pour distinguer les néoplasmes bénins des néoplasmes malins : une étude clinicopathologique et immunophénotypique de 100 cas. Am J Surg Pathol. 2002; 26(5):551-566. doi :10.1097%2F00000478-200205000-00002.
- Pacak K, Wimalawansa SJ. Phéochromocytome et paragangliome. Endocr Pract. 2015; 21(4):406-412. doi :10.4158/EP14481. RA.
- Goffredo P, Sosa JA, Roman SA. Phéochromocytome malin et paragangliome : une analyse au niveau de la population de la survie à long terme sur deux décennies. J Surg Oncol. 2013; 107(6):659-664. doi :10.1002/jso.23297.
- Li J, Yang CH. Amélioration de la prise en charge préopératoire chez les patients atteints de phéochromocytome surrénalien. Int J Clin Exp Med. 2014; 7(12):5541-5546. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4307515.
- Chen H, Sippel RS, O’Dorisio MS, Vinik AI, Lloyd RV, Pacak K ; Société nord-américaine des tumeurs neuroendocrines (NANETS). Les lignes directrices consensuelles de la North American Neuroendocrine Tumor Society pour le diagnostic et la prise en charge des tumeurs neuroendocrines : phéochromocytome, paragangliome et cancer médullaire de la thyroïde. Pancréas. 2010; 39(6):775-783. doi :10.1097/MPA.0b013e3181ebb4f0.
- Sutton MG, Sheps SG, Lie JT. Prévalence du phéochromocytome cliniquement insoupçonné : examen d’une série d’autopsies de 50 ans. Mayo Clin Proc. 1981; 56(6):354-360.
- Lairmore TC, Ball DW, Baylin SB, Wells SA Jr. Prise en charge des phéochromocytomes chez les patients atteints de syndromes multiples de néoplasie endocrinienne de type 2. Ann Surg. 1993; 217(6):595-603. doi :10.1097/00000658-199306000-00001.
- Étendue de l’surrénalectomie pour le néoplasme surrénalien : épargne corticale (sous-totale) versus surrénalectomie totale. Surg Clin North Am. 2004; 84(3):743-753. doi :10.1016/j.suc.2004.01.003.
- Renard J, Clerici T, Licker M, Triponez F. Phéochromocytome et paragangliome abdominal. J Visc Surg. 2011; 148(6) :E409 à E416. doi :10.1016/j.jviscsurg.2011.07.003.
- Irvin GL 3e, Fishman LM, Sher JA. Phéochromocytome familial. Chirurgie. 1983; 94(6):938-940. https://www.surgjournal.com/article/0039-6060(83)90403-8/texte intégral.
- Mercan S, Seven R, Ozarmagan S, Tezelman S. Surrénalectomie rétropéritonéale endoscopique. Chirurgie. 1995; 118(6):1071-1076. doi :10.1016/S0039-6060(05)80116-3.
- Alesina PF, Hinrichs J, Meier B, Schmid KW, Neumann HP, Walz MK. Chirurgie mini-invasive de conservation corticale pour les phéochromocytomes bilatéraux. Langenbecks Arch Surg. 2012; 397(2):233-238. doi :10.1007/s00423-011-0851-2.
Cite this article
Brown TC, Carling T. Surrénalectomie rétropéritonéoscopique postérieure bilatérale avec épargne corticale du côté droit. J Med Insight. 2021; 2021(282). doi :10.24296/jomi/282.

