Bilateral Posterior Retroperitoneoscopic Adrenalectomy with Cortical Sparing on Right Side
Main Text
Table of Contents
Cortical-sparing adrenalectomy allows for resection of adrenal tumor(s) while preserving unaffected adrenal tissue to prevent adrenal insufficiency. This is especially important in patients affected with bilateral adrenal tumors, typically pheochromocytomas.
Posterior retroperitoneoscopic adrenalectomy (PRA) allows for a minimally invasive approach to adrenal gland resection compared with the more traditional laparoscopic transabdominal adrenalectomy and open approaches. The PRA technique is increasingly used by high-volume endocrine surgeons throughout the world. This approach is ideal to address patients with bilateral disease and was used in this case of a patient presenting with bilateral pheochromocytomas in the setting of multiple endocrine neoplasia 2A syndrome.
Posterior retroperitoneoscopic adrenalectomy (PRA) was first popularized in Germany by Walz and colleagues. The adrenal glands are accessed via a retroperitoneal approach using laparoscopic instrumentation and CO2 insufflation.1 In doing so, the surgeon avoids entry into the peritoneal cavity and mobilization of surrounding viscera, including bowel, liver, spleen, and pancreas. Compared with open and laparoscopic transabdominal adrenalectomy (LTA) approaches, this technique promotes patient recovery with decreased length of stay, less pain, and reduced risk of ileus.2-4
One of the advantages of PRA is that it enables bilateral access to both adrenal glands via a minimally invasive approach without requiring patient repositioning during the operations.2 Patients presenting with bilateral adrenal tumors, typically pheochromocytomas due to Von Hippel Lindau (VHL) or multiple endocrine neoplasia type 2 (MEN2) syndrome, are ideal candidates for this approach. In both disease processes, bilateral tumors frequently occur. As such, patients may require bilateral adrenalectomies to achieve a biochemical cure.
In order to prevent postoperative acute adrenal failure (Addisonian crisis), cortical-sparing adrenalectomy can be performed. During this procedure, the culprit tumor tissue is removed while preserving normal adrenal tissue.5 Traditionally, this technique has been described alongside open and LTA approaches; however, for bilateral disease, the PRA approach is increasingly being utilized with success.
The patient is a 31-year-old female who presented with biochemically unequivocal bilateral pheochromocytomas. She had symptoms of hypertension and palpitations, which prompted further investigation by her primary care doctor. Her laboratory workup was significant for elevated free plasma metanephrines at 642 pg/ml (reference range < 57 pg/mL) and normetanephrine at 2284 pg/ml (reference < 148 pg/ml) as well as elevated urinary metanephrines, consistent with a pheochromocytoma. Cross-sectional imaging included a CT of the abdomen with IV contrast. The CT revealed bilateral adrenal nodules with precontrast Hounsfield units and overall imaging characteristics of bilateral pheochromocytoma (Figure 1). A close review of her imaging demonstrated normal-appearing adrenal cortex tissue on the right side (Figure 2) that would be conducive to cortical-sparing adrenalectomy on this side.
Because of her young age and the presence of bilateral tumors, she was investigated further for other MEN2A-associated tumors, specifically medullary thyroid cancer and primary hyperparathyroidism. She was indeed found to have an elevated serum calcitonin level of 229 pg/ml (reference < 5 pg/ml), and ultrasound and fine-needle aspiration findings were consistent with medullary thyroid carcinoma of the right thyroid lobe. Her serum intact parathyroid hormone and calcium levels were unremarkable.
CT and MRI are the primary radiologic techniques used for imaging normal and abnormal adrenal glands. On CT, pheochromocytomas are often well defined from the surrounding tissues and typically demonstrate precontrast Hounsfield units of 30–40. Smaller lesions tend to appear simple and solid, while larger lesions may have more cystic characteristics as well as other complex features. On MRI, pheochromocytomas have a classic “light-bulb” appearance on T2 weighted images. Functional imaging may also be obtained, especially when metastatic disease is a concern. The most common method is use of 131 I- and 123 I-metaiodobenzylguanidine (MIBG), which is an analogue of norepinephrine and localizes preferentially to sympathomedullary tissues.6
We prefer the patient to undergo either adrenal protocol CT or MRI within approximately 3–6 months of the operation for operative planning. In this case, the patient was referred to us after having a CT showing bilateral adrenal masses with imaging characteristics suspicious for pheochromocytoma. In a detailed review or her imaging, it was evident that the inferior medial limb of the right adrenal gland had unaffected adrenal cortex tissue that could be potentially preserved during resection.
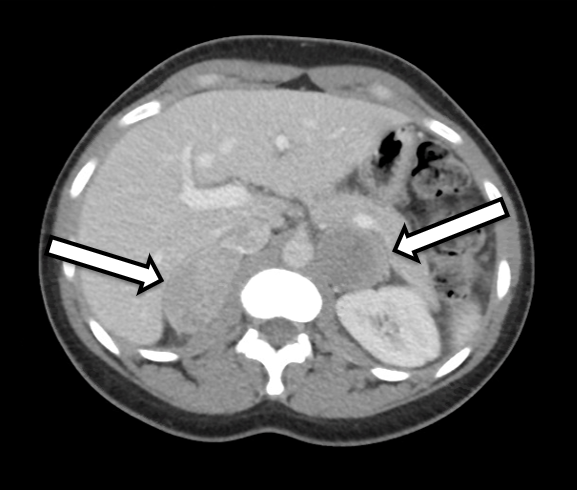
Figure 1. Transverse view of nodule within right thyroid lobe;
white arrows denote microcalcifications.
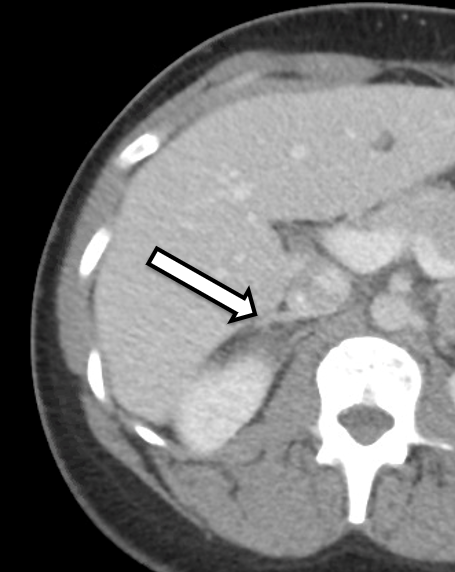
Figure 2. Enhanced view of CT of the abdomen demonstrating
normal adrenal cortex (arrow) of the right adrenal
gland that was preserved.
Pheochromocytomas originate from neural crest cells of the adrenal medulla and secrete excess amounts of catecholamines. The overall prevalence of pheochromocytomas has been estimated between 1:6500 and 1:2500, with an average age of onset between 40–50 years and a slightly higher prevalence in women. Most tumors are benign.6 Interestingly, there is no single pathological characteristic, including size, mitotic rate, or vascular or capsular invasion, that can accurately predict malignant potential, though various predictive algorithms have been created.7 Approximately 10–15% of tumors can present with and/or develop metastatic disease, indicative of malignant transformation.8 Disease-specific survival for malignant pheochromocytomas has been estimated to be approximately 70% at 5 years based on analysis of the Surveillance, Epidemiology, and End Results (SEER) database.9
Standard treatment for pheochromocytomas is surgical resection. Prior to the operation, the patient must undergo catecholamine blockade, which is usually performed using the nonselective alpha-receptor blocker phenoxybenzamine or a selective alpha-blocker such as doxazosin. Additional antihypertensive agents may be needed as well, including beta-blockers. However, beta-blockers should only be started after a patient has been placed on alpha-blockers to prevent a hypertensive crisis caused by an unopposed beta-receptor blockade. Furthermore, patients are chronically dehydrated and require preoperative fluid resuscitation.10
Patients afflicted with pheochromocytomas frequently suffer episodes of severe hypertension and other clinical manifestation of excessive catecholamine production, including palpitations, headaches, panic attacks, and diaphoresis.11 While blood pressure medications may provide partial relief, the only long-term, durable treatment is resection of the culprit lesion(s). Furthermore, those who continue without intervention are at risk of severe hypertensive crises, potentially leading to death.12 Therefore, patients with pheochromocytomas should seek surgical treatment expeditiously.
In patients presenting with bilateral pheochromocytomas, cortical-sparing adrenalectomy can prevent the occurrence of adrenal insufficiency while resecting the culprit tissue. Previous studies have shown that total adrenalectomy can be highly morbid. For example, in a series published by Lairmore et al. in which 43 patients underwent complete adrenalectomy for bilateral pheochromocytomas, 23% suffered episodes of adrenal insufficiency, and one patient died from an Addisonian crisis.13 Furthermore, patients who have undergone total adrenalectomy report poor quality of life and frequent hospitalizations related to adrenal insufficiency.14
Cortical-sparing adrenalectomy is an invaluable option for patients with genetic syndromes associated with pheochromocytomas. These syndromes include MEN2, VHL, and neurofibromatosis type 1 (NF1), as well as others. It is important to note that patients diagnosed with bilateral pheochromocytomas should be referred for genetic testing. Calcitonin testing may be a helpful adjunct to identify patients with MEN2, as genetic testing can take several months to be performed and properly interpreted. Approximately 40–80% of patients with either MEN2A or VHL will develop bilateral pheochromocytomas, and these tumors are usually benign.15 As such, cortical-sparing adrenalectomy can remove affected tissue, while leaving behind enough tissue to prevent adrenal insufficiency with minimal risk of recurrence.
The first case of cortical-sparing adrenalectomy was described by Irvin et al. in 1983.16 Since then, some institutions have reported success in performing cortical-sparing adrenalectomy through either open or minimally invasive approaches with fairly low recurrence rates, and importantly, low occurrence of adrenal insufficiency. 14 The use of PRA has been increasing as high-volume endocrine surgeons have implemented this technique. PRA was first described in 1995 and then further developed through the experience of Walz and his colleagues.1, 4, 17 Retrospective studies comparing PRA to LTA show decreased operative times, decreased blood loss, and no difference in long-term outcomes.
More recently, PRA is being utilized for cortical-sparing adrenalectomy as well. In a recent series, the experience over a 25-year period of performing cortical-sparing adrenalectomies for bilateral pheochromocytomas using the RPA approach was described. Sixty-six patients were operated on with a total of 101 adrenalectomies performed. Their mean operative time was 128 minutes for bilateral surgeries, and they reported only two major complications. They were able to perform a cortical-sparing operation in 89 of the cases and of those patients, 91% did not require steroids postoperatively. Only one patient had persistent disease, while no recurrences were reported.18 Patients should be cautioned that they might have some adrenal insufficiency following surgery. This is dependent on the size of the remnant adrenal tissue and the preservation of blood flow. Temporary adrenal insufficiency is managed in close consultation with endocrinology and appropriate laboratory testing.
The procedure is performed under general anesthesia with endotracheal intubation. Due to the hemodynamic alterations that can occur during surgery due to the tumor, an arterial line is placed for blood pressure monitoring, and the anesthesiologist administers vasoactive drugs as needed. A Foley catheter is placed to monitor urinary output. Depending on the degree of catecholamine elevation and other patient factors, a central venous line can be placed for additional venous access and monitoring of central venous pressure.
After induction of general anesthesia and endotracheal intubation, the patient is placed in a prone jackknife position with the hips bent at 90 degrees. A Cloward table with a Cloward Surgical Saddle is used to allow the abdomen to hang in a dependent fashion. The jackknife position and the Cloward Surgical Saddle open the retroperitoneal space to maximize exposure. All pressure points, including the face, arms, hips, and legs, are extensively padded. The iliac crest, the tips of the 11th and 12th ribs, and the edge of the paraspinous muscles are important landmarks marked by the surgeon. The initial incision is placed just inferior to the tip of the 12th rib. Scissors are used to sharply divide the soft tissue and enter the retroperitoneum. The surgeon’s finger is then used to bluntly clear space and guide placement of a 5-mm port medially and laterally, both angled at about 30 degrees and aimed toward the position of the adrenal gland. A 10-mm balloon port is then placed in the middle incision. The retroperitoneum is insufflated with CO2 through high-flow tubing to an insufflation pressure of 25 mmHg.
A 5-mm 30-degree scope is inserted in the 10-mm port, and a LigaSure device is used to create the retroperitoneal space. The dissection begins by identifying the paraspinous muscles medially, which is then followed by the identification of the kidney inferiorly. The camera is then typically moved to the medial port and the surgeon uses a LigaSure device and a bowel grasper through the lateral and central ports, respectively. Dissection continues over the superior pole of the kidney and along the paraspinal muscles medially, towards the direction of the adrenal gland. Exposure is facilitated in part by downward pressure on the kidney. As the adrenal gland is identified, the inferior extent of the gland and its relationship to the adrenal vein is determined. On the right, this dissection reveals the IVC, which the adrenal gland is dissected off of to reveal the adrenal vein. On the left, the adrenal vein originates from the left renal vein.
At this point, the precise relationship of the unaltered cortical adrenal tissue to the culprit tumor is determined. This is in part determined by a detailed evaluation of preoperative imaging in conjunction with intraoperative findings. Occasionally, intraoperative ultrasound may be useful. Once a suitable plane is identified, the pheochromocytoma is then divided from the normal adrenal tissue with a LigaSure device. The adrenal gland is a highly vascularized, so meticulous attention is given to hemostasis as the gland is transected. In this case, the inferior medial limb of the native right adrenal gland was suitable for preservation. If possible, the vein should be preserved. However, this not necessary, nor is it always feasible, given that there is additional venous drainage along the small adrenal arteries. During the current operation, the adrenal vein was found to be directly entering the tumor and as such it was divided with clips. At this point, the medial and lateral attachments of the tumor are taken down, leaving the superior attachment to allow the tumor to hang down and provide a superior site of counter-traction. Care is taken not to enter the peritoneal cavity during the lateral and cephalad dissections. Once the tumor has been circumferentially dissected, the superior attachments are taken, and the gland is removed from the retroperitoneal space with an Endocatch device. The 10-mm port is closed in layers, while the 5-mm port sites are closed only at the level of the skin.
Final pathology revealed a 5.2-cm right pheochromocytoma and a 5.6-cm left pheochromocytoma. Testing performed on postoperative day one demonstrated mildly decreased cortisol production. As such, the patient was temporarily placed on a low dose of oral steroids. The patient was discharged home on postoperative day two with excellent recovery when she was seen in the clinic two weeks following her operation, with a plan to wean off the low dose of prednisone.
Andrew frame, Cloward Surgical Saddle, LigaSure device, and Endocatch retrieval bag.
The authors have no disclosures.
The patient referred to in this video article has given their informed consent to be filmed and is aware that information and images will be published online.
Citations
- Walz MK, Peitgen K, Hoermann R, Giebler RM, Mann K, Eigler FW. Posterior retroperitoneoscopy as a new minimally invasive approach for adrenalectomy: results of 30 adrenalectomies in 27 patients. World J Surg. 1996;20(7):769-774. doi:10.1007/s002689900117.
- Callender GG, Kennamer DL, Grubbs EG, Lee JE, Evans DB, Perrier ND. Posterior retroperitoneoscopic adrenalectomy. Adv Surg. 2009;43(1):147-157. doi:10.1016/j.yasu.2009.02.017.
- Perrier ND, Kennamer DL, Bao R, et al. Posterior retroperitoneoscopic adrenalectomy: preferred technique for removal of benign tumors and isolated metastases. Ann Surg. 2008;248(4):666-674. doi:10.1097/SLA.0b013e31818a1d2a.
- Walz MK, Peitgen K, Walz MV, et al. Posterior retroperitoneoscopic adrenalectomy: lessons learned within five years. World J Surg. 2001;25(6):728-734. doi:10.1007/s00268-001-0023-6.
- Lee JE, Curley SA, Gagel RF, Evans DB, Hickey RC. Cortical-sparing adrenalectomy for patients with bilateral pheochromocytoma. Surgery. 1996;120(6):1064-1071. doi:10.1016/S0039-6060(96)80056-0.
- Farrugia FA, Martikos G, Tzanetis P, et al. Pheochromocytoma, diagnosis and treatment: review of the literature. Endocr Regul. 2017;51(3):168-181. doi:10.1515/enr-2017-0018.
- Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26(5):551-566. doi:10.1097%2F00000478-200205000-00002.
- Pacak K, Wimalawansa SJ. Pheochromocytoma and paraganglioma. Endocr Pract. 2015;21(4):406-412. doi:10.4158/EP14481.RA.
- Goffredo P, Sosa JA, Roman SA. Malignant pheochromocytoma and paraganglioma: a population level analysis of long-term survival over two decades. J Surg Oncol. 2013;107(6):659-664. doi:10.1002/jso.23297.
- Li J, Yang CH. Improvement of preoperative management in patients with adrenal pheochromocytoma. Int J Clin Exp Med. 2014;7(12):5541-5546. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4307515.
- Chen H, Sippel RS, O'Dorisio MS, Vinik AI, Lloyd RV, Pacak K; North American Neuroendocrine Tumor Society (NANETS). The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39(6):775-783. doi:10.1097/MPA.0b013e3181ebb4f0.
- Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma: review of a 50-year autopsy series. Mayo Clin Proc. 1981;56(6):354-360.
- Lairmore TC, Ball DW, Baylin SB, Wells SA Jr. Management of pheochromocytomas in patients with multiple endocrine neoplasia type 2 syndromes. Ann Surg. 1993;217(6):595-603. doi:10.1097/00000658-199306000-00001.
- Walz MK. Extent of adrenalectomy for adrenal neoplasm: cortical sparing (subtotal) versus total adrenalectomy. Surg Clin North Am. 2004;84(3):743-753. doi:10.1016/j.suc.2004.01.003.
- Renard J, Clerici T, Licker M, Triponez F. Pheochromocytoma and abdominal paraganglioma. J Visc Surg. 2011;148(6):e409-e416. doi:10.1016/j.jviscsurg.2011.07.003.
- Irvin GL 3rd, Fishman LM, Sher JA. Familial pheochromocytoma. Surgery. 1983;94(6):938-940. https://www.surgjournal.com/article/0039-6060(83)90403-8/fulltext.
- Mercan S, Seven R, Ozarmagan S, Tezelman S. Endoscopic retroperitoneal adrenalectomy. Surgery. 1995;118(6):1071-1076. doi:10.1016/S0039-6060(05)80116-3.
- Alesina PF, Hinrichs J, Meier B, Schmid KW, Neumann HP, Walz MK. Minimally invasive cortical-sparing surgery for bilateral pheochromocytomas. Langenbecks Arch Surg. 2012;397(2):233-238. doi:10.1007/s00423-011-0851-2.
Cite this article
Brown TC, Carling T. Bilateral posterior retroperitoneoscopic adrenalectomy with cortical sparing on right side. J Med Insight. 2021;2021(282). doi:10.24296/jomi/282.